Compliance Gaps in Clinical Trial Disclosure
Article
Regulatory & Compliance

Disclosure compliance is often not a knowledge problem. It is a visibility problem.
Noncompliance surfaces during an internal audit, an inspection, or an article critical of transparency practices, and at that point, the question isn't what went wrong; it’s who was responsible?
There are over 80 countries with clinical data disclosure requirements, and often the problem is these disclosure obligations are tracked by people rather than systems. Someone knew the requirements. Someone updated the trial. Someone else made a judgment call. But when something changed, whether it was a regulation, a trial adding a site location in a new country, or even just a team member, the trial fell between the cracks, and the data were not disclosed properly.
Disclosure compliance is often not a knowledge problem. Since the information is distributed across departments, affiliates, and contract research organizations (CROs), it becomes a visibility problem. The rules change more often than internal processes can easily capture in a standard operating procedure (SOP), and the portfolio of trials may be large, making it time consuming to identify trials that require disclosure work.
It may be obvious, but focusing on submitting data to a few leading registries, like ClinicalTrials.gov in the US, CTIS in the EU, or jRCT in Japan, is not the same as being globally compliant with requirements in over 80 countries.

Most disclosure teams working in industry have rock-solid processes for submission to the top two or three registries; they track statuses, meet deadlines, and update registry records. It works today because that is where the attention and investments have been in the past decade.
When that process lives in someone's head or in a spreadsheet, it works until it fails silently or is disrupted whenever a team member leaves.
Recently, companies have come to realize that since they are responsible for compliance with regulations globally, they need a centralized approach to tracking disclosure obligations and compliance for each country; they cannot rely solely on their network of affiliates and CROs to own that for them. Although disclosure execution can be local, content coordination and compliance oversight should be a centralized function.
Centralization means evaluating each trial against a complex set of criteria, requiring over 70 pieces of information about the trial to determine in which countries and on which registries the data must be disclosed, when the data are due, and when the data must be updated. And then rerunning the assessments every time the trial data or regulations change.
While clinical trial data disclosure requirements started as regulations, they rapidly expanded well beyond the regulatory domain, which exerted a strong pressure for increased transparency. As disclosure practices have evolved over the past 25+ years, many sponsors have developed SOPs and policies that establish a two-tier disclosure process. The first tier is based on satisfying the disclosure regulations and represents the minimum compliance standard. The second tier addresses the additional transparency pressures and may have tighter deadlines and a broader scope than is strictly required by the regulations.

Regulations establish the legal baseline for what must be disclosed, where, and by when. These requirements are subject to inspections and may carry significant regulatory penalties. Non-regulatory requirements captured in company policies are tested in internal audits and may be assessed by external researchers and industry critics.
If your company is only tracking compliance against the regulatory tier, while you might be legally compliant, you will likely not catch issues with the expanded commitments. For example, in the USA, the protocol must be registered no later than 21 days after study start; however, to comply with the Helsinki Accords or to submit the trial data to a medical journal, the study must be registered before the first patient is enrolled.
These two compliance standards must be tracked separately so sponsors can confirm that each trial disclosure complied with regulations in 80+ countries and dozens of non-regulatory requirements.
During an inspection, the sponsor must be able to prove that they have fully complied with regulations. Additionally, internal audits should assess compliance with corporate policies, as these reflect public commitments to patients, investigators, and the public. Not only is this necessary to manage the corporate reputation, but it is also necessary because failing to meet some of these non-regulatory requirements can have consequences, such as being unable to publish the data in a recognized medical journal or the ethics committee refusing to authorize the trial.
I've seen organizations where the transparency policy evolved independently of the tracking reports that verify compliance, resulting in an incomplete understanding of whether commitments are met globally. When the two tiers aren't tracked separately, a sponsor who was compliant with both may be unable to demonstrate it. As auditors always tell me: “If it’s not documented, it didn’t happen.”
When a CRO or service provider manages disclosure tasks on local registries, it is tempting to rely on their expertise and assume that everything is completed in compliance with regulations.
However, the obligation to comply with the requirements and the liability for failures remain with the sponsor and do not transfer to these partners. While vendors may have the expertise and processes to submit data to the trial registries, it is the sponsor that will have to defend an audit.
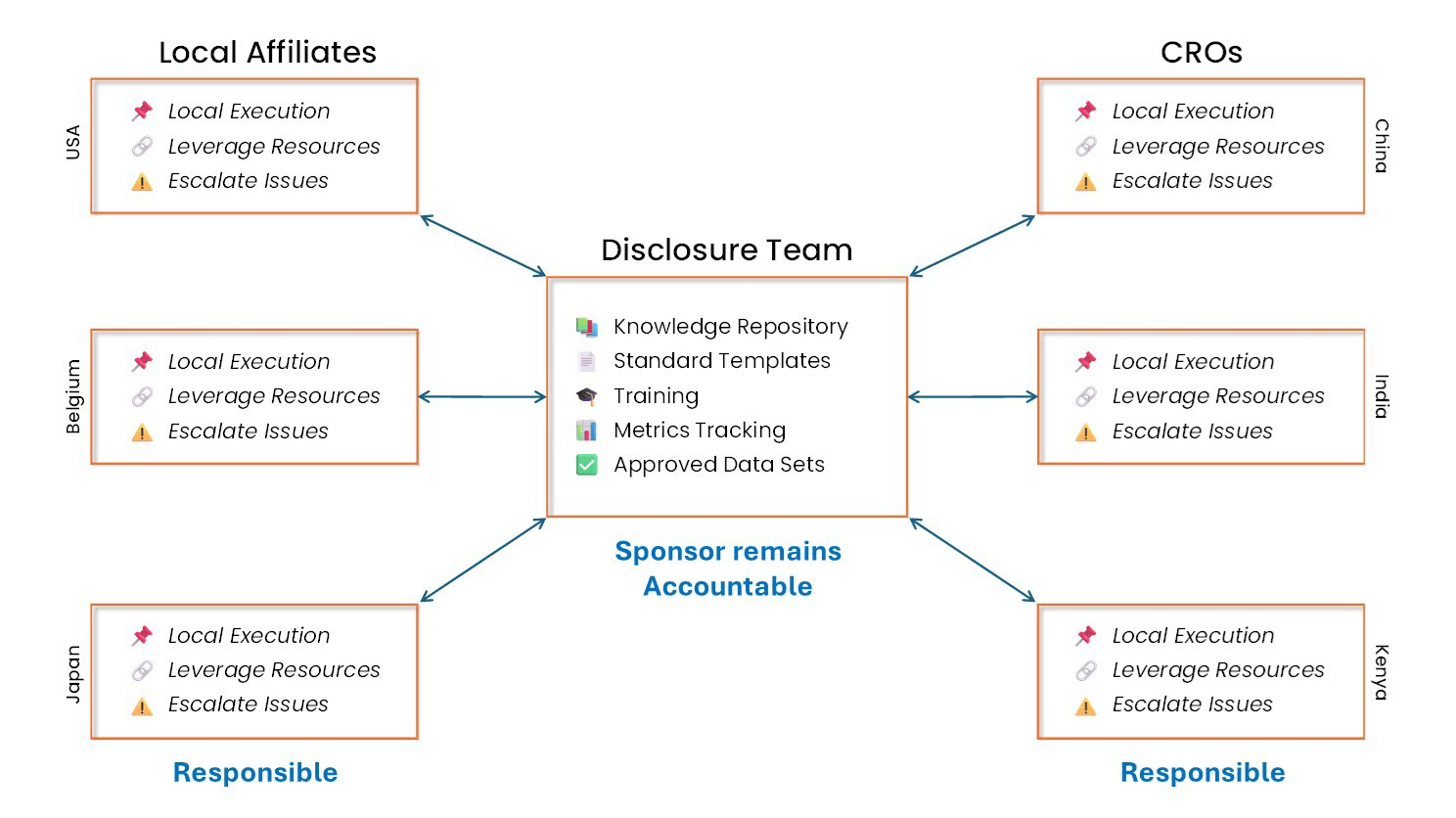
Sponsors should always know what was disclosed, on which registry, and by when; it is not enough to trust that someone else did it. Beyond managing global compliance, there are at least three additional reasons to coordinate disclosure centrally:
While distributed disclosure execution is practical, distributed accountability poses a significant risk.
Disclosure teams are typically confident they know the requirements for the registries they support directly. However, the team is often not sure they are up to date on the regulations governing local registries. This means they can’t really be sure whether the CROs managing those submissions are compliant.
To assess your current approach, here are a few questions you should be able to answer for your organization:
Although many teams have deep expertise for the registries they cover, the challenge is whether the systems and processes can manage a global trial portfolio, where the requirements are constantly changing.
To keep up with the increasing complexity of global disclosure, start with a centrally managed regulatory knowledge base to track requirements for every country in which your organization conducts trials.
To ensure timely submissions, the best practice is to use a decision tree to encode the requirements for each country, which can then be used to determine which trials must be disclosed, on which registries, and by what deadlines.
When disclosure requirements change, teams face two challenges:
The first burden can be tricky. Teams document what they know and research what they don’t yet fully understand, but the challenge is to become aware of regulatory changes that aren't widely announced. The sponsor community is quick to share new requirements and discuss the implications of changes in jurisdictions such as the US, the EMA, the UK, and Japan. However, new requirements in countries where fewer trials are conducted often go unnoticed, leading to either a mad dash to assess updates or noncompliance. Still, most teams have evolved a process for handling it, whether it's a subscription service, a regulatory affairs colleague monitoring national authority and registry websites in all countries where the sponsor is conducting trials, or an alert from a CRO.
The second burden arises once it is clear that a regulatory change affects clinical trial disclosure requirements. Now the question is: Which trials in your portfolio are impacted by the change?
Whether the change makes disclosure mandatory where it was previously optional, requires updates to the registration after initial submission, or changes timelines and scope, the challenge is identifying all trials that now require additional disclosures. Especially for organizations with large trial portfolios or those that outsource research to CROs, this can mean a significant effort.
For example, before June 2022, trials with sites in Kenya could be registered on the Pharmacy and Poisons Board (PPB), but it was voluntary. As of June 8, 2022, trial registration has become mandatory. This change required sponsors to have a process for monitoring disclosure regulations in Kenya and then identify all trials with sites in Kenya that met the revised requirements.
These two burdens are separate problems that require separate answers, even if they are closely related. Trial sponsors need a reliable source of current requirements and a system to apply those requirements to every trial whenever anything changes.
Most disclosure teams are aware of the problems this article has described, including the many local requirements evolving more rapidly than they can track reliably, and trial assessments that are managed in spreadsheets and rely on the interpretation of one or two experts. Recognizing these issues is the first step, but the challenge then is how to start fixing them.
In my experience, organizations that have shifted to a global model were successful when they built capabilities in sequence rather than trying to address everything at once. This article about the CLARITY model, Citeline’s seven-step framework for balancing global compliance with local autonomy, describes such a model.
Two core components of a successful shift to global disclosure coordination are a centrally maintained library of requirements and a systematic way to determine which trials are affected. The library of local requirements is the regulatory intelligence component, providing an understanding of what disclosure is required in each country where you conduct trials, with the necessary level of detail to support accurate trial assessments. This is where tools like TrialScope Intelligence add value: replacing manual monitoring across dozens of jurisdictions with a curated, continuously updated knowledge base.
The second component is the daily assessment of all trials based on the requirements in the regulatory intelligence library, determining what must be disclosed, on which registry, and by when. The TrialScope Disclose global registry compliance module does this by applying decision logic across 60+ registries and distinguishing between regulatory obligations and policy commitments.
Intelligence without execution is research, and the knowledge does not reliably determine disclosure obligation. Without regulatory intelligence, execution is guesswork, and existing processes and systems rely on incomplete or outdated information to assess transparency mandates.
Why is tracking disclosure obligations across 80+ countries so difficult?
Disclosure compliance is primarily a visibility problem, not a knowledge problem. Requirements are distributed across departments, affiliates, and CROs, and regulations change more frequently than internal SOPs can capture. Relying on people rather than systems means knowledge walks out the door when team members leave.
Can a sponsor transfer accountability to a CRO or service provider managing local registries?
No. The obligation to comply and the liability for failures remain with the sponsor regardless of which partner executes the submission. Sponsors must always know what was disclosed, on which registry, and by when. There are additional reasons to maintain central oversight: all disclosed data are public and could affect patent filings, disclosure dates can impact IP strategy, and global consistency requires that the same trial tells the same story across all registries.
What are the two main challenges when a disclosure regulation changes?
The first challenge is simply becoming aware of the change and correctly interpreting it. The second challenge is identifying which trials in your portfolio are affected by the change.
What are the foundational components of a global disclosure compliance model?
Two core components are essential. The first is a centrally maintained regulatory knowledge base covering requirements for every country where trials are conducted, continuously updated to reflect changes across 60+ registries, replacing manual monitoring. The second is a systematic daily assessment process that applies those requirements to each trial, determining what must be disclosed, on which registry, and by when.




Don’t let the April 28 enforcement catch you off guard. Gain critical insights and practical guidance to achieve compliance with new UK clinical trial transparency rules for trial registration and results publication.

