TrialScope EXTRA 2025: Smart Innovation for Disclosure’s Future
Article
Regulatory & Compliance

Large and small biopharma companies alike are taking a more global, coordinated approach to clinical trial disclosure as requirements grow in scope. Citeline’s annual TrialScope EXTRA event in New York City for TrialScope Disclose customers provided a look back at the past year’s accomplishments and a look ahead at future innovations.
 Jaclyn Spedaliere
Jaclyn Spedaliere
The theme of this year’s TrialScope EXTRA clinical trial disclosure event in New York City was future-proofing disclosure demands through smart innovation, and the presentations emphasized Citeline’s efforts in this regard. Jaclyn Spedaliere, VP, Regulatory Sales, opened TrialScope EXTRA 2025 with a quick recap of the changes to Citeline’s regulatory offerings over the past year, including the Core Data form for results, an expansion of TrialScope Intelligence’s library of clinical trial requirements, and further integration and enhancement of TrialScope Disclose, Citeline’s comprehensive solution for clinical trial disclosure management.
She also referenced the relaunch of the TrialScope ECHO program, which fosters open dialogue between customers and product teams, gathering actionable feedback on product usability, performance, and features. “This is very important internally for us to have this feedback loop so that we can continue to innovate and develop our software,” she said.
Thomas Wicks, Head of Transparency Operations, spoke about how the disclosure market is evolving and how Citeline is responding to the shifting trends. He observed that both large and small biopharma companies are taking a more global, coordinated approach to disclosure as requirements grow in scope.
“If you’re disclosing everything everywhere all at once, then you need to do earlier planning for registration and results, you need to think about the plain language summaries, you’re doing a lot more coordination across the organization,” he said.

Wicks outlined the new regulations that have taken effect during the past year, including updates to the Declaration of Helsinki that require all human-based trials to be registered and disclosed, and all results to be disclosed to participants. This trend toward earlier-phase trial disclosures could also be seen in updates to the US Food and Drug Administration (FDA) Good Clinical Practice (GCP) guidelines and EU CTR rules. “If it’s in humans, if it’s an interventional study, it’s getting registered and results are disclosed.”
He also emphasized the increasing push toward — and importance of — plain language summaries (PLS).
Regulations are constantly changing, making it tough for regulatory and disclosure teams to keep up. Over 120 disclosure-impacting new documents have been published in the last 12 months, but over one-third of those are already obsolete, Wicks said. “Who’s going to have a full-time staff for checking 196 countries’ worth of regulations?”
If disclosure isn’t done, or done right, it can have profound consequences. Wicks said he knows of three patents that have been rejected because of disclosure problems. “Inspections have started. No one has been fined yet, but it will happen.”
According to Wicks, artificial intelligence (AI) has promise in disclosure through intelligent data ingestion that reduces manual entry burden. Well-structured, standards-aligned data can dramatically improve AI utility, he noted.
Right now, uses for AI include drafting PLS tailored to patient needs and conducting conformance checks to flag inconsistencies with registry requirements before submission. “It’s really about working smarter, faster, and with greater confidence.”
However, Wicks said there need to be guardrails in place when incorporating AI into disclosure procedures. He cited positivity bias as one area that is sometimes overlooked. “Without using some kind of agents to check for it, it will tend to take a very positive spin on what’s going on in your trial, which is unacceptable. It’s considered promotional language. So you have to be really looking out for that.”
If you ever wondered about the machinations behind the vast amount of data behind Citeline’s TrialScope Intelligence, wonder no more. Disclosure Regulations Research Manager Ben Evans gave attendees a behind-the-scenes glimpse at the solution that is a repository of global clinical trial disclosure regulations.
Evans noted that registration requirements “have transformed into a complex web of global obligations that vary significantly by region. Different countries have developed their own disclosure regulations, all varying in scope, timelines, and registry requirements. Disclosure obligations have also broadened considerably; requirements have moved beyond basic registration to comprehensive reporting that includes plain language summaries, and each of these elements requires specialized knowledge as well as timing consideration. Tracking these evolving regulations is, without a doubt, resource intensive.”
He noted that the consequences for noncompliance also have been escalated, including financial penalties and even criminal liability. Evans referenced a recent white paper he collaborated on with Francine Lane, Vice President of Global Transparency, exploring the gap between non-disclosure penalties and their actual enforcement. Belgium has the most stringent penalties with a fine of €500,000 and three years in prison, although this has not yet been enforced. He said it is mainly social accountability, not legislation, that is driving compliance globally.
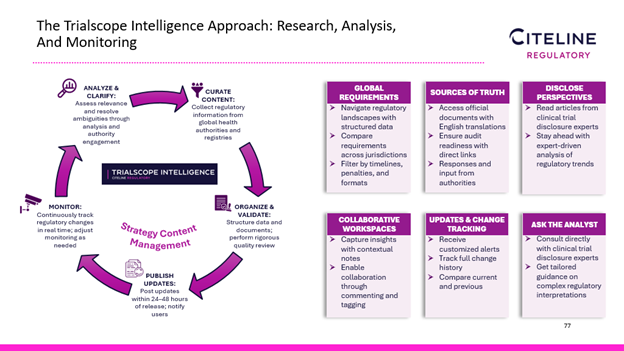
Citeline’s response to these regulatory disclosure challenges, Evans said, is the TrialScope Intelligence solution. He said it uses a lifecycle approach incorporating research, analysis, synthesis, and monitoring. TrialScope Intelligence now covers 193 countries, with 75 requirement records, and 60 registry records, all of which are constantly being updated. In addition, there are 950 source documents, all with English translations and direct links to the source materials.
Evans said the solution has intuitive features:
In short, he said, TrialScope Intelligence stays one step ahead of the regulatory radar.
In this session, Evans was joined by Sarah Stark, head of clinical trial transparency at Astellas. They focused on recent updates and regulatory trends identified by TrialScope Intelligence, as well as best practices and practical with real-world implementation strategies.

Sarah Stark, Astellas, and Ben Evans, Citeline
Evans discussed the challenges inherent with protocol registration. “Protocol registration is one thing,” he said, “but the timing for the protocol registration is another.” Stark said that, for Astellas, the use of a regulatory intelligence tool is necessary to manage the complexity of protocol registration requirements across different jurisdictions. Prior to implementing TrialScope Intelligence, she said Astellas had created its own Excel spreadsheet with registration requirements for all the countries it was working with plus the activity required to complete the registration, whether it was a standalone database, clinical trial application, or an ethics committee submission or approval activity. In addition, Astellas lists the requirements, the database, who's responsible, what activity populates that registry, as well as the due date.

Stark noted that Astellas disclosure policy goes above and beyond country requirements and regulations. In managing disclosure, she said CRO contracts are another ongoing challenge. Study teams typically want to end the CRO contracts as quickly as possible. She said meeting the 12-month timeline for most results is not an issue; however, due to a great deal of turnover, the study team members who were involved at the trial’s start might not be there when results are due six to 12 months later.
Evans and Stark also discussed the unique submission requirements associated with pediatric trials, moving on to tackle the topic of growing requirements for PLS. Stark said Astellas has been posting PLS in the public domain since 2014.
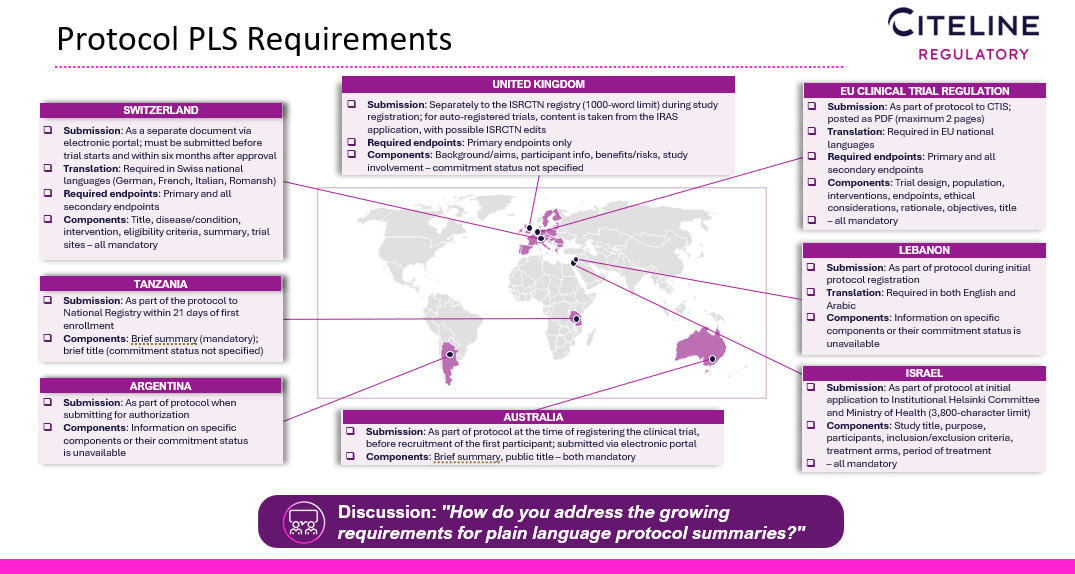
In addition, Astellas has been translating studies occurring in China or Japan. She added that it would be helpful if regulatory authorities provided more standardization and at least specific titles or sections that need to be included.
“I think change is the only stability in transparency and probably in the pharma industry,” Stark said. “We definitely try to stay ahead of the curve.”
Lane highlighted the disclosure achievements of Citeline’s clients over the past year, noting that they have registered 424 new studies and updated nearly 3,000 studies on ClinicalTrials.gov alone.
Lane gave a peek into the figures for Citeline’s Trial Summaries portal, the world’s largest source for PLS. She observed that there are 2,877 studies listed in Trial Summaries, and nearly 1,700 of them have PLS available. Over 17,000 PLS documents have been downloaded from TrialSummaries.com, and people from 165 countries have come to the website to look at the studies there, she said.
Lane also noted the updates and enhancements that have been made to TrialScope Disclose, including performance improvements, optimized loading times for forms, and added more options for downloading study information. For Trial Summaries, improvements include disclaimer enhancements that allow users to define the disclaimer content for both yes and no options, single sign on for users, and a QR code for use when sharing study results. And for TrialScope Intelligence, there have been layout improvements, navigation enhancements, new fields, and rich formatting in executive summaries.
 Arran Carter
Arran Carter
Product Manager Arran Carter demonstrated the new Core Data results form. He noted that Core Data allows users to submit to multiple supported registries using one form, saving time and eliminating duplication. In fact, using Core Data slashes the total number of potential registry fields to be filled by up to 50%.
When showing the attendees the new form, Carter said, “You immediately see the power of Core Data” because one field can cover multiple registries, such as CTgov, CTIS (the EU registry), and jRCT (Japan’s registry). He said Core Data accepts Japanese for jRCT and languages will be expanded as more registries are added.
The form shows which sections have been approved, pending, or rejected, as well as which information will be made public in the registries. When edits are made, the form tracks them, and if an edit is made to a section of the form that’s already approved, the approval will be removed. “It’s very intuitive, very easy to see,” Carter said.
Other features of the Core Data results form include a section for custom content that allows the customer to add additional fields to the form as necessary and the ability to add a non-system approver to the form.
Brian Kuzich, Senior Solution Consultant, and Sahil Khatter, Principal Project Manager, held an interactive session on ways companies benchmark disclosure performance. Some methods cited by the attendees include the National Institutes of Health (NIH) acceptance rate, Good Pharma Scorecard criteria, and company-specific measures such as how many results have been disclosed and making sure Phase I studies are posted on CTgov.
 L-R: Sahil Khatter, Brian Kuzich
L-R: Sahil Khatter, Brian Kuzich
One attendee who helped develop the Good Pharma Scorecard criteria spoke. She found that companies with poor scores made excuses for why the criteria were wrong. Even though the scorecard is one of the more collaborative ratings systems, companies tend to have their own metrics, she said.
Many attendees said their companies expand beyond existing disclosure rules, including publishing early-phase trials and disclosing protocols as soon as they become finalized. In some cases, however, there was significant pushback, such as from early-phase teams who didn’t want their trials to be made public.
When asked how benchmarking can be improved, some attendees suggested better benchmarking criteria for smaller, local registries and clearer guidelines on what can’t be made public in PLS.
Challenges were the theme of the day as Lane moderated a fireside chat on the challenges around PLS and the benefits of using a centralized site like TrialSummaries.com. She was joined by Maureen Mooney Kashuba, Director of Medical Writing & Disclosure at Merck, and Julie Holtzople, President of Holtzople Consulting. Lane presented questions she gathered from other sponsors and from conversations she has had across the industry on this topic.

L-R: Merck’s Maureen Mooney Kashuba, consultant Julie Holtzople, and Citeline’s Francine Lane
“Let’s start with one,” Lane said, “that goes straight to the heart of the matter: overcoming legal and regulatory concerns when sharing PLS on a public site like TrialSummaries.com, especially since it’s not directly required by regulation.”
Kashuba shared how she initially received pushback at Merck: ”When we first explored sharing PLS publicly, the initial response from legal and regulatory teams was ‘no’ — sometimes a very firm ‘no.’ There was nervousness because there was no regulation explicitly requiring it. But I kept pushing.”
She noted that early attempts to distribute printed summaries through sites failed; sites did not want to do the mailing, they weren’t tracking addresses, and it was not sustainable. “TrialSummaries.com gave us an electronic, scalable solution” she said, reminding attendees that TrialSummaries.com is sponsor-agnostic. “Someone once said it’s like the ClinicalTrials.gov of plain language summaries.”
Lane said some academic sponsors have questioned whether sharing PLS without explicit informed consent language violates patient rights and asked the panelists to weigh in on that. Holtzople replied: “My understanding is this: Informed consent governs direct communication with participants. If you’re not reaching out directly — just posting online — it’s not a problem. Yes, it’s easier if the consent form includes language like ‘You may be informed of the results,’ but retroactively posting summaries doesn’t violate consent. The key difference is whether you’re actively contacting participants. That’s where you need prior permission.”
Next, Lane asked the panelists if they are exploring formats beyond PDFs, such as audio summaries, comics, or videos. Holtzople recommends sponsors explore this with caution. “Audio makes sense, especially in countries with lower literacy rates or for people with visual impairments. But video and images raise concerns — legal and compliance teams worry about promotional tone. Even a cartoon character’s smile can be seen as biased.”
When asked how they are spreading the word — among sites, participants, and the general public — Kashuba said, “We’ve piloted end-of-study letters for investigators with optional participant letters, reminding them about the availability of summaries. But it’s hard — budgets, time, and engagement drop once the study ends.”
Holtzople added that real change will require modifying processes and even site contracts. “It’s about culture shift,” she said.
Regarding the FDA’s 2024 GCP guidelines, Kashuba said, “The guidance now expects investigators to inform participants which treatment arm they were in and share results — preferably in plain language. ... There’s a real opportunity to use TrialSummaries.com to support compliance here.”
Holtzople agreed, adding, “And don’t forget the 2024 update to the Declaration of Helsinki — it adds pressure to return results in a timely, complete, and understandable way.”
Beyond regulations, sharing PLS helps boost a sponsor’s brand reputation. “It’s not just about compliance,” Holtzople said. “It’s about showing that we care, that we’re investing in transparency. It helps build trust in research — globally.”
 Suzanne Caruso
Suzanne Caruso
As far as AI goes, Suzanne Caruso, General Manager, Clinical & Regulatory, said the industry is demanding more integration of AI. “The industry is trying products with AI integrated, but they are not always trusting of the output initially,”she said.
No matter how AI is used, Caruso said, subject matter experts will still be necessary. “I want to be really clear on this. … [S]ubject matter expertise needs to sit on top of everything that’s happening.”
One specific area where there is a lot of demand is AI agents, a bespoke tool that can take a task and run it for users so they don’t have to. Caruso says Citeline will be looking at agents to see how they can help customers take on different tasks.
When considering where AI is headed next, Caruso said more companies are looking to incorporate AI in Phases I–III of clinical development. Many companies don’t think they’re spending enough on AI, and “that gives me hope,” she said. “I want people who are excited to be learning more and spending more in this because I think it will bring efficiencies across the entire clinical trials spectrum.”



To enable the booking feature, please enable all cookies in your browser.


